Our Research
 During earlier studies on the signalling pathways that control phototaxis and thermotaxis in Dictyostelium, we discovered that these behaviours are highly sensitive to genetic defects affecting the mitochondria. Mitochondrial disease often produces central nervous system defects and mitochondrial dysfunction is a common feature of most neurodegenerative diseases, including Alzheimer's, Parkinson's, Motor Neuron and Huntington's Diseases, so our initial accidental creation of mitochondrial disease in Dictyostelium led us to establish Dictyostelium strains with each of these diseases or other neurodegenerative diseases including the lysosomal disorders, Mucolipidosis and Batten Disease.
During earlier studies on the signalling pathways that control phototaxis and thermotaxis in Dictyostelium, we discovered that these behaviours are highly sensitive to genetic defects affecting the mitochondria. Mitochondrial disease often produces central nervous system defects and mitochondrial dysfunction is a common feature of most neurodegenerative diseases, including Alzheimer's, Parkinson's, Motor Neuron and Huntington's Diseases, so our initial accidental creation of mitochondrial disease in Dictyostelium led us to establish Dictyostelium strains with each of these diseases or other neurodegenerative diseases including the lysosomal disorders, Mucolipidosis and Batten Disease.
We are investigating the signal transduction pathways in slug behaviour using a combination of pharmacological, genetic, cell physiological and molecular biological approaches. Our recent work revealed a protein signalling complex for phototaxis in which the participating proteins are assembled on a scaffold provided by a protein called filamin which binds to the actin skeleton of the cell.
 We discovered that signal transduction for phototaxis and thermotaxis in slugs is more sensitive to the presence of mitochondrial defects than other cellular activities such as growth and division. Thus phototaxis and thermotaxis are impaired by mitochondrial mutations created by plasmid insertions in a minority of the mitochondrial genomes in the cell. The same defects are observed when the folding of proteins in the mitochondria is impaired by antisense inhibition of the expression of chaperonin 60. Chaperonin 60 is encoded on a nuclear gene and is required for the proper folding of proteins in the mitochondria. Undersupply of chaperonin 60 therefore causes serious mitochondrial disease. The severity of the undersupply caused by antisense inhibition is determined by the number of copies of the antisense inhibition construct and this is different in every every cell line carrying the construct. This allows the generation of genetic dose-response curves relating phenotype ("symptoms") to the severity of the underlying genetic defect. In addition to the phototaxis and thermotaxis defects, mitochondrial disease in Dictyostelium causes slow growth (without affecting the rate of uptake of nutrients by phagocytosis or pinocytosis), a misdirection of cells into the stalk differentiation (programmed cell death) pathway, and less efficient aggregation
We discovered that signal transduction for phototaxis and thermotaxis in slugs is more sensitive to the presence of mitochondrial defects than other cellular activities such as growth and division. Thus phototaxis and thermotaxis are impaired by mitochondrial mutations created by plasmid insertions in a minority of the mitochondrial genomes in the cell. The same defects are observed when the folding of proteins in the mitochondria is impaired by antisense inhibition of the expression of chaperonin 60. Chaperonin 60 is encoded on a nuclear gene and is required for the proper folding of proteins in the mitochondria. Undersupply of chaperonin 60 therefore causes serious mitochondrial disease. The severity of the undersupply caused by antisense inhibition is determined by the number of copies of the antisense inhibition construct and this is different in every every cell line carrying the construct. This allows the generation of genetic dose-response curves relating phenotype ("symptoms") to the severity of the underlying genetic defect. In addition to the phototaxis and thermotaxis defects, mitochondrial disease in Dictyostelium causes slow growth (without affecting the rate of uptake of nutrients by phagocytosis or pinocytosis), a misdirection of cells into the stalk differentiation (programmed cell death) pathway, and less efficient aggregation
Unexpectedly we found that these defects associated with mitochondrial dysfunction are a result of chronic activation of the cellular energy-sensing alarm protein AMPK. The Dictyostelium mitochondrial disease model thus suggests that the complex pathology of human mitochondrial disease might be explained partially by chronic AMPK signalling rather than an energy insufficiency per se. This discovery resulted in my being awarded the Australasian Science Prize for 2007 and provided a completely new perspective on how mitochondrial dysfunction damages cells. You can hear more about this discovery in an interview conducted by Dr. Moira Gunn on May 7th, 2007 in a BioTech Nation radio broadcast of September 28th, 2007 and made available on line by ITConversations. It has since been confirmed that AMPK activities are abnormally chronically elevated in Alzheimer's, Huntington's and Motor Neuron Diseases and at least in Huntington's Disease this contributes to the pathological outcomes.
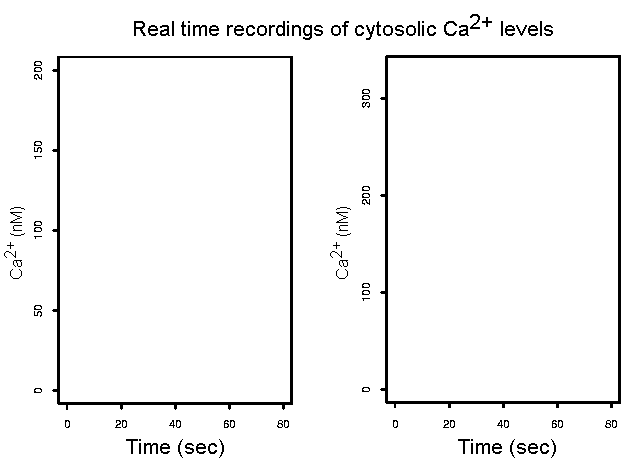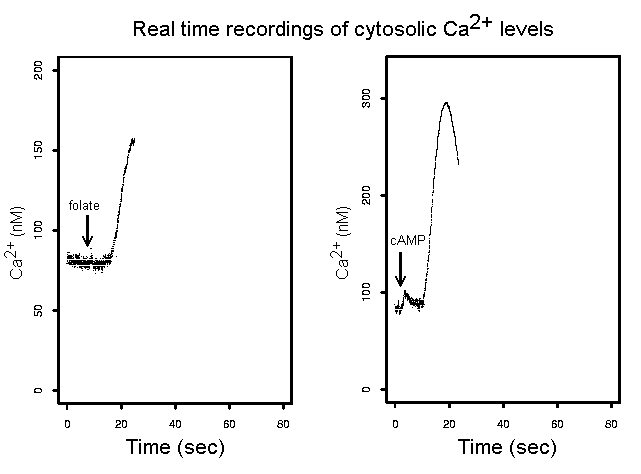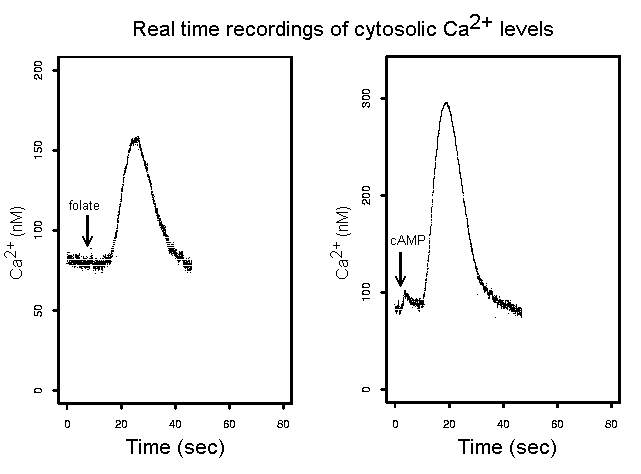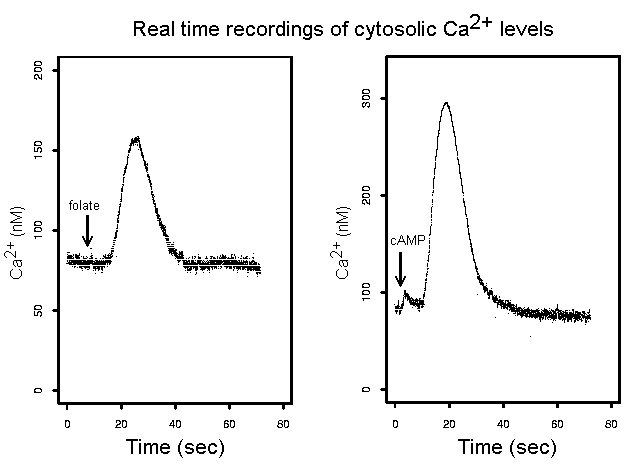 The third major project in the laboratory concerns intracellular Ca2+ signals. Calcium signals in cells are partly regulated by the mitochondria and can be abnormal in cells with mitochondrial dysfunction. They also play a central role in neuronal function so we are studying their dysregulation in mitochondrial and neurodegenerative diseases. Using an assay based on expression in Dictyostelium of recombinant aequorin, a Ca2+-sensitive luminescent protein, we are able to measure cytosolic Ca2+ concentrations in a population of cells every 20 msecs, down to concentrations of about 25 nM to within a few nM. Using this assay we measure intracellular Ca2+ signals initiated by various extracellular stimuli including the morphogen DIF and the chemoattractants cAMP and folic acid. We are investigating the roles played by abnormal Ca2+ signalling in mitochondrial and neurodegenerative diseases.
The third major project in the laboratory concerns intracellular Ca2+ signals. Calcium signals in cells are partly regulated by the mitochondria and can be abnormal in cells with mitochondrial dysfunction. They also play a central role in neuronal function so we are studying their dysregulation in mitochondrial and neurodegenerative diseases. Using an assay based on expression in Dictyostelium of recombinant aequorin, a Ca2+-sensitive luminescent protein, we are able to measure cytosolic Ca2+ concentrations in a population of cells every 20 msecs, down to concentrations of about 25 nM to within a few nM. Using this assay we measure intracellular Ca2+ signals initiated by various extracellular stimuli including the morphogen DIF and the chemoattractants cAMP and folic acid. We are investigating the roles played by abnormal Ca2+ signalling in mitochondrial and neurodegenerative diseases.